Cancer is a hyperproliferative disorder that involves transformation, dysregulation of apoptosis, proliferation, invasion, angiogenesis and metastasis.
Extensive research during the last 30 years has revealed much about the biology of cancer. Drugs used to treat most cancers are those that can block cell signaling, including growth factor signalling (e.g., epidermal growth factor); prostaglandin production (e.g., COX-2); inflammation (e.g., inflammatory
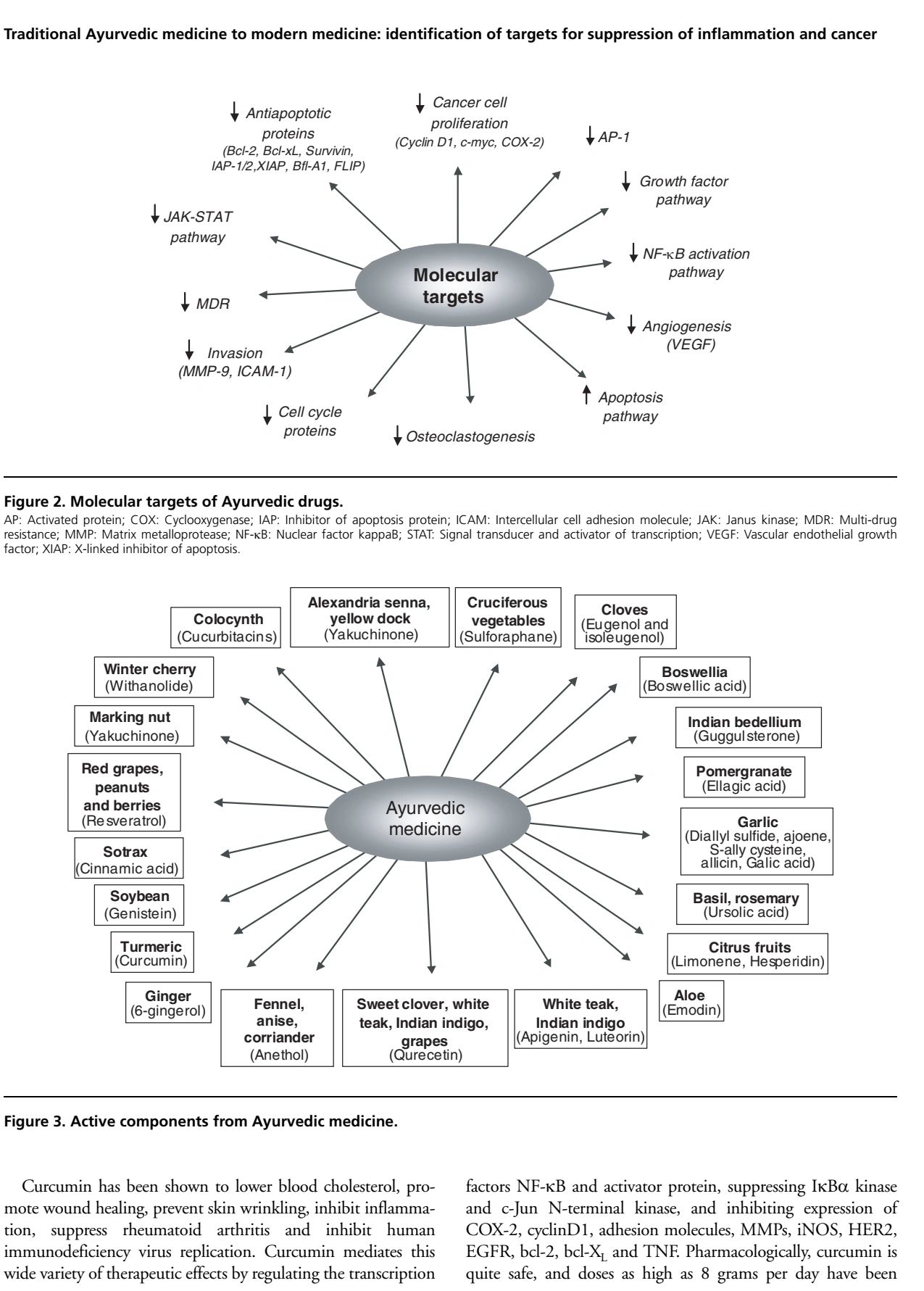
cytokines: NF-κB, TNF, IL-1, IL-6, chemokines); drug resistance gene products (e.g., multi-drug resistance); cell cycle proteins (e.g., cyclin D1 and cyclin E); angiogenesis (e.g., vascular endothelial growth factor); invasion (e.g., matrix metalloproteinases); antiapoptosis (e.g., bcl-2, bcl-XL, XIAP, survivin, FLIP); and cellular proliferation (e.g., c-myc, AP-1, growth factors). Numerous reports have suggested that Ayurvedic plants and their components mediate their effects by modulating several of these recently identified therapeutic targets.
However, Ayurvedic medicine requires rediscovery in light of our current knowledge of allopathic (modern) medicine. The focus of this review is to elucidate the Ayurvedic concept of cancer, including its classification, causes, pathogenesis and prevention; surgical removal of tumours; herbal
remedies; dietary modifications; and spiritual treatments.


Introduction
According to the International Agency for Research on Cancer (IARC), in 2002, cancer killed > 6.7 million people around the world; another 10.9 million new cases were diagnosed; and at the current rate, an estimated 15 million people will be diagnosed annually by 2020. Cancer is one of the leading causes of death in the US and around the world. Several chemotherapeutic, cytotoxic, and immunomodulating agents are available in Western medicine to treat cancer. Besides being enormously expensive, these drugs are associated with severe side effects and morbidity. Still, the search continues for an ideal treatment that has minimal side effects and is cost-effective. Today, in Western medicine, only a limited number of plant products are being used to treat cancer. However, some of the widely used anticancer drugs, such as taxol and vinca alkaloids, are obtained from medicinal plants. This review focuses on the ancient perspective of cancer and how it can be integrated with modern science for the best treatment of cancer (Figure 1). Ayurveda, one of the significant traditional forms of medical practice in India, has produced many useful leads in developing medications for chronic diseases. Almost 25 centuries ago, Hippocrates proclaimed, ‘Let food be thy medicine and medicine be thy food.’ According to a recent report by Newman et al., as many as 65% of formally synthetic hypertension drugs are plant-based [1]. Of the 121 prescription drugs in use today for cancer treatment, 90 are derived from plants. Almost 74% of these, including taxol, was discovered by investigating a folklore claim [2,3]. Between 1981 and 2002, 48 out of 65 drugs approved for cancer treatment were natural products, based on natural products, or mimicked natural products in one form or another [1]. These phytochemicals are commonly called chemotherapeutic or chemopreventive agents.
Phytochemicals may fight disease through suppression of the inflammatory response. Dysregulated inflammation contributes to many diseases, including cancer [4,5]. It stands to reason then, that suppression of inflammation, whether by phytochemicals or other means, should delay the onset of disease [2,3]. Tumourigenesis is a multistep process that begins with cellular transformation, advances towards hyperproliferation, and culminates in the acquisition of invasive potential and angiogenic properties and the establishment of metastatic lesions [6]. This process can be activated by any of the various environmental carcinogens (such as cigarette smoke, industrial emissions, gasoline vapors), inflammatory agents (such as TNF and H 2O2), tumefaction promoters (such as phorbol esters and okadaic acid). This multistep process of carcinogenesis involves three phases: tumor initiation, promotion, and progression. Several population-based studies indicate that people in Southeast Asian countries have a much lower risk of developing colon, gastrointestinal, prostate, breast, and other cancers when compared with their Western counterparts.
It is likely that dietary constituents, such as garlic, ginger, soy, curcumin, onion, tomatoes, cruciferous vegetables, chilies, and green tea, play an essential role in protection from these cancers. These dietary agents are considered to halt the transformative, hyperproliferative, and inflammatory processes that initiate carcinogenesis. Their inhibitory properties may ultimately suppress the final steps of carcinogenesis as well, namely angiogenesis and metastasis.
These dietary constituents have been classified as chemopreventive agents, and their capacity to delay the onset of carcinogenesis has been studied extensively.
Because these chemopreventive agents are derived from natural sources, they are considered pharmacologically safe. The current review, although brief, evaluates the untapped therapeutic potential of these agents in the setting of several molecular targets that are currently under investigation.
1. Role of the NF-κB activation pathway in tumourigenesis
NF-κB is a family of closely related protein dimers that bind to a common sequence motif in DNA called the κB site [7]. The molecular identification of its p50 subunit (v-REL) as a member of the reticuloendotheliosis (REL) family of viruses provided the first evidence that NF-κB is linked to cancer. Research over the past decade has revealed that NF-κB is an inducible transcription factor for genes involved in cell survival, cell adhesion, inflammation, differentiation, and growth. In most resting cells, NF-κB is sequestered in the cytoplasm by binding to the inhibitory IκB proteins that block the nuclear localization sequences of NF-κB. NF-κB is activated by a variety of stimuli, such as carcinogens, inflammatory agents, and tumor promoters, including cigarette smoke, phorbol esters, okadaic acid, H2O2, and TNF. These stimuli promote dissociation of IκBα through phosphorylation, ubiquitinylation, and its ultimate degradation in the proteasomes. This process unmasks the nuclear localization sequence of NF-κB, facilitating its nuclear entry, binding to κB regulatory elements, and activation of transcription of target genes. Many of the target genes that are activated are critical to the establishment of the early and late stages of aggressive cancers, including expression of cyclin D1, apoptosis suppressor proteins such as bcl-2 and bcl-XL and those required for metastasis and angiogenesis, such as matrix metalloproteases (MMPs) and vascular endothelial growth factor (VEGF).
2. Role of the AP-1 activation pathway in cancer prevention
Activated protein-1 (AP-1) is another transcription factor that regulates the expression of several genes involved in cell differentiation and proliferation. Functional activation of the AP-1 transcription complex is implicated in tumor promotion as well as in malignant transformation. This complex consists of either homo- or heterodimers of the constituents of the JUN and FOS family of proteins [8]. This AP-1-mediated transcription of several target genes also can be activated by a complex network of signaling pathways that involve external signals such as growth factors, mitogen-activated protein kinases (MAPKs), extracellular signal-regulated protein kinases and c-jun N-terminal kinase (JNK). Some of the target genes activated by the AP-1 transcription complex mirror those activated by NF-κB and include cyclin D1, bcl-2, bcl-XL, VEGF, MMP, and urokinase plasminogen activator (uPA). Expression of genes such as MMP, and especially uPA, promotes angiogenesis and invasive growth of cancer cells. Most importantly, AP-1 can also improve the transition of tumor cells from an epithelial to a mesenchymal morphology, one of the early steps in tumor metastasis. These oncogenic properties of AP-1 are primarily dictated by the dimer composition of the AP-1 family proteins and their post-transcriptional and translational modifications.
3. Role of proliferation and apoptosis in tumourigenesis
Several reports have been published in the past eight years showing that activation of NF-κB promotes cell survival and proliferation, and downregulation of NF-κB sensitizes the cells to apoptosis. The mechanism through which NF-κB helps this proliferation, and cell survival mechanisms have become increasingly apparent. Expression of several genes, including bcl-2, bcl-XL, inhibitor-of-apoptosis protein (IAP), survivin, cyclin D1, TNF receptor-associated factor 1(TRAF1), and TRAF2, has been reported to be upregulated by NF-κB [9]. The proteins coded by these genes function primarily by blocking the apoptosis pathway. Several studies have demonstrated that NF-κB activation promotes cell survival and proliferation mechanisms and that suppression of NF-κB leads to abrogation of these mechanisms. Similarly, c-JUN is primarily a positive regulator of cell proliferation because c-jun-deficient fibroblasts have a marked proliferation defect in vitro and in vivo. c-jun protein, once fully activated by JNK kinases, induces transcription of the positive regulators of cell cycle progression, such as cyclin D1, and represses the negative regulators, such as the tumor suppressor p53 and the cyclin-dependent kinase inhibitor p16 (INK4A). Moreover, activated and oncogenic AP-1 can antagonize apoptosis in several tumors.
4. Growth factor activation pathway in tumourigenesis
The potent cell proliferation signals generated by various growth factor receptors, such as the epidermal growth factor receptor, insulin-like growth factor-1 receptor, and VEGF receptor networks, constitute the basis for receptor-driven tumorigenicity in the progression of several cancers [6]. The consequence of these abnormal growth factor receptor signaling pathways include increased cell proliferation, suppression of apoptotic signals (especially under anchorage-independent conditions), and an increase in the tumor’s invasive behavior, which contributes to metastatic spread and the growth of new blood vessels. Several chemopreventive phytochemicals, including curcumin, genistein, resveratrol, and catechins, recently are powerful inhibitors of several growth factor receptors, including epidermal growth factor receptor (EGFR). Some of these phytochemicals, such as curcumin, also can inhibit the ligand-stimulated activation of the EGFR, indicating that they have the potential to break the autocrine loops that are established in several advanced cancers [10]. The inhibitory actions of these phytochemicals have several other potential advantages in treating patients with late-stage cancers. A blockade of EGFR, for an example may predispose the cancer cells to apoptosis.
Moreover, inhibition of EGFR disables the protein’s capacity to provide the cancer cell the matrix-independent survival support it needs to expand and acquire invasive potential. Third, these chemopreventive chemicals function by inhibiting other tyrosine kinases, such as c-src, that are involved in the activation of the G-protein-coupled receptor to the transactivation of EGFR occurs extensively in established cancers. Finally, most of these phytochemicals also inhibit, by a similar mechanism, the HER2/neu receptor, which is overexpressed in breast, prostate, ovarian, and lung cancers. Curcumin has been shown not only to inhibit the tyrosine kinase activity of this receptor but also to deplete the protein itself. It does so by interfering with the function of the ATP-dependent GRP94 chaperone protein, which is involved in maintaining the properly folded state of the receptor [11].
Moreover, by inhibiting HER2/neu, most of these phytochemicals also can interfere with the cross-talk between the receptor and the estrogen receptor pathways in these cancers. Thus, they may be beneficial in treating hormone-resistant breast cancer patients by restoring their hormone responsiveness.
5. Role of the JAK–STAT pathway in tumourigenesis
Although cancer arises through several genetic or epigenetic mechanisms that contribute to several abnormal oncogenic signaling pathways, all seem to converge on a minimal number of nuclear transcription factors that function as final effectors, triggering specific gene expression patterns for particular cancer. These belong to the canonical signal transducers and activators of transcription (STAT) family of proteins [12]. They can be activated by phosphorylation through janus kinase (JAK) or cytokine receptors, G-protein-coupled receptors or growth factor receptors (such as EGFR); by platelet-derived growth factor receptors that have intrinsic tyrosine kinase activity; or by intracellular nonreceptor tyrosine kinase recruitment. Of the seven STAT proteins identified so far constitutive activations of STAT3 and STAT5 have been
implicated in multiple myeloma, lymphomas, leukemias and
several solid tumors, making these proteins logical targets for
cancer therapy. These STAT proteins contribute to cell survival and growth by preventing apoptosis through increased
expression of anti-apoptotic proteins, such as bcl-2 and
bcl-X
L. Recently, STAT 3 was shown to be a direct activator
of the VEGF gene, which is responsible for increased angiogenesis. More importantly, the increased expression of
STAT3 and STAT5 transcription factors are crucially
involved in the processes through which tumors evade
immunological surveillance by increasing the feeling of
immune-suppressing factors and decreasing the expression
of pro-inflammatory cytokines that are responsible for the
maturation of the dendritic cells [13].
6. Role of multi-drug resistance in tumourigenesis
MDR in human cancer is often associated with overexpression of the mdr-1 gene, which encodes a 170 kDa transmembrane protein, termed P-glycoprotein (P-gp). P-glycoprotein is considered to be of prognostic relevance in different tumor types. It is involved in resistance to natural product-based chemotherapeutics, including taxanes, anthracyclines, vinca alkaloids, podophyllotoxins, and camptothecins. Although several reports suggest that P-170 is clinically relevant in hematological malignancies, its role in solid tumors is not well understood. Its overexpression is correlated with the poor outcome observed in patients treated with chemotherapy and presenting drug resistance. Activation of the MDR-1 gene or selection of intrinsically MDR neoplastic cells may occur at the early stages of tumourigenesis of oral cancers before the real evidence of cellular transformation [14]. Thus, contact with possible chemical carcinogens, such as those of tobacco smoke, may induce activation of the MDR-1 gene. MDR-1 product expression in oral squamous cell carcinoma might suggest that overexpression of this protein could constitute a hallmark of potential more aggressive phenotype for this type of neoplasia. Quantitative flow-cytometric analysis of P-GP expression showed a significant increase in P-gp levels in untreated primary oral tumors and dysplastic lesions as compared with healthy oral tissues. A marked considerable improvement in P-GP expression was observed in recurrent oral carcinomas as compared with standard oral tissues and dysplastic lesions. Among recurrent tumors, a significant increase in the level of P-gp was observed in T4-stage tumors as compared with T3-stage tumors. Thus, P-gp is differentially expressed during oral tumourigenesis and might be an indicator of the biological behavior of oral malignancies [15]. Activation of MDR-related gene expression also occurs during the tumourigenesis of urothelial cancers and that it may confer de novo and acquired drug resistance on urothelial carcinomas [16]. Like cytochrome P450s (CYP3A4), P-gp is vulnerable to inhibition, activation, or induction by herbal constituents.
7. Role of COX-2 in tumourigenesis
Numerous preclinical studies point to the importance of regulating cyclooxygenase-2 (COX-2) expression in the prevention and, most importantly, to the treatment of several malignancies. This enzyme is overexpressed in practically every premalignant and malignant condition involving the colon, liver, pancreas, breast, lung, bladder, skin, stomach, head and neck, and esophagus [17]. COX-2 overexpression is a consequence of the deregulation of transcriptional and post-transcriptional control. Several growth factors, cytokines, oncogenes, and tumor promoters, stimulate COX-2 transcription. Expression of COX-2 is increased in HER2/neu-expressing breast carcinomas owing to enhanced ras signaling. Depending upon the stimulus and the cell type, different transcription factors, including AP-1, NF-IL-6, and NF-κB, can stimulate COX-2 transcription [17]. Wild-type p53 protein expression can suppress COX-2 transcription, whereas the mutant p53 protein cannot. Consistent with this observation, increased COX-2 levels are seen in several epithelial cancers that express mutant p53. Taken together, these findings suggest that the balance between the activation of oncogenes and the inactivation of tumor suppressor genes and the expression of several pro-inflammatory cytokines can modulate the expression of COX-2 in tumors. Complicating matters further is the fact that conventional cancer therapies, such as radiation and chemotherapy, can induce COX-2 and prostaglandin biosynthesis. Thus, inhibition of this enhanced COX-2 activity in tumors has therapeutic potential.
8. Role of angiogenesis in tumourigenesis
Angiogenesis, the regulated formation of new blood vessels from existing ones, is the basis of several physiological processes, such as embryonic development, placenta formation, and wound healing. It is one of the best examples of how a tumor can take control of these processes and deregulate them to its advantage. In the regular and orderly formation of new blood vessels, the endothelial cell receives the stimulatory signal and secretes MMP and heparanase, which causes the extracellular matrix to dissolve. The tight junction between the endothelial cells is then altered, and the cells project through the newly created space where freshly formed endothelial cells organize into fresh capillary tubes. This allows the sprouting vessel to grow toward the source of fresh blood [18]. When a tumor tries to build new blood vessels, most of these standard physiological rules governing new blood vessel growth are subverted. Blood vessels newly formed by tumors often have incomplete basement membranes, and the microvasculature is often chaotic, following convoluted paths without organization. These vessels also have a disproportionate ratio of endothelial cells to pericytes and abnormal pericyte coverage. The new blood vessels are hyperpermeable because of an imbalance of pro- and antiangiogenic factors, and they are often leaky [18]. Moreover, tumor cells themselves try to mimic the properties of endothelial cells and form a loose vasculogenic meshwork by processes such as vessel cooption and vasculogenic mimicry [19]. Thus, interference with the mechanisms of angiogenic switch, vessel cooption , and the vasculogenic imitation will be of high therapeutic value in several advanced cancers.
9. Role of cyclins in tumourigenesis
Hundreds of types of cancer exhibit global changes in gene expression, but only a minimal number of crucial alterations are common to all tumors. These common alterations are related to those that disrupt the normal cell cycle control checkpoints. The retinoblastoma and tumor suppressor p53 proteins that are crucial for these controls are usually lost in several cancers. The central role of the G1 to S and the G2 to M transitions and the corresponding checkpoints in cancer development are well established [20]. Formation and regulation of enzyme complexes with the D-type cyclins and their partners and the B-type cyclins with their associated proteins are particularly well characterized, as is the control of retinoblastoma function by phosphorylation.
1. Guggulsterone (Commiphora mukul)
1. Guggulsterone (Commiphora mukul)
1. Guggulsterone (Commiphora mukul)

Guggulsterone [4,17(20)-pregnadiene-3,16-dione] is a plant sterol derived from the gum resin (Guggulu) of the tree Commiphora Mukul. The resin has been used in Ayurvedic medicine for centuries to treat a variety of ailments, including obesity, bone fractures, arthritis, inflammation, cardiovascular disease, and lipid disorders [28,29]. The antiarthritic and anti-inflammatory activities of gum guggul were demonstrated as early as 1960 by Gujral et al. [30]. Sharma et al. showed guggul’s activity in experimental arthritis induced by a mycobacterial adjuvant [31]. The effectiveness of guggul for treating osteoarthritis of the knee also has been demonstrated [32]. Recent studies have shown that guggulsterone is an antagonist for the bile acid receptor farnesoid X receptor [33,34]. Other studies have shown that guggulsterone enhances transcription of the bile salt export pump [35], thereby regulating cholesterol homeostasis. An understanding of the molecular mechanisms underlying guggulsterone is just now emerging. In 2003, Meselhy et al. showed that guggulsterone could suppress inflammation by inhibiting inducible nitric oxide synthetase (iNOS) expression induced by lipopolysaccharide in macrophages [36]. Because most inflammatory diseases are mediated through the activation of NF-κB, a nuclear transcription factor [7,37], the authors hypothesize that it is involved in guggulsterone’s activity. Guggulsterone suppresses DNA binding of NF-κB induced by TNF, phorbol ester, okadaic acid, cigarette smoke condensate, hydrogen peroxide, and IL-1. Guggulsterone also suppressed the constitutive NF-κB activation expressed in most tumor cells. Besides, guggulsterone decreases the expression of gene products involved in antiapoptosis (IAP1), X chromosome-linked IAP, Bfl-1/A1, bcl-2, cFLIP and survivin), proliferative genes (cyclin D1, c-myc), and metastatic genes (MMP-9, COX-2, and VEGF). This correlated with the enhanced apoptosis induced by TNF and chemotherapeutic agents [38].
2. Curcumin (Curcuma longa)
1. Guggulsterone (Commiphora mukul)
1. Guggulsterone (Commiphora mukul)
Curcumin (diferuloylmethane) is an active component of turmeric (Curcuma longa), which has been used as a spice and as an Ayurvedic medicine for centuries on the Indian subcontinent. Curcumin has been shown to suppress carcinogenesis of the skin, liver, lung, colon, stomach, and breast. It has also been shown to inhibit the proliferation of a wide variety of tumor cells in culture and to promote apoptosis through Bid cleavage, cytochrome c release, caspase-9 activation and then caspase-3 activation [39-60]. Curcumin has been shown to lower blood cholesterol, promote wound healing, prevent skin wrinkling, inhibit inflammation, suppress rheumatoid arthritis, and inhibit human immunodeficiency virus replication. Curcumin mediates this wide variety of therapeutic effects by regulating the transcription factors NF-κB and activator protein, suppressing IκBα kinase and c-Jun N-terminal kinase, and inhibiting expression of COX-2, cyclinD1, adhesion molecules, MMPs, iNOS, HER2, EGFR, bcl-2, bcl-XL, and TNF. Pharmacologically, curcumin is quite safe, and doses as high as 8 grams per day have been administered orally to humans with no side effects. The numerous therapeutic activities of curcumin, its pharmacological safety, and its color qualify it as ‘Indian solid gold.’ Extensive research over the last 50 years has indicated that curcumin can both prevent and treat cancer. Curcumin’s anti-cancer potential stems from its ability to suppress the proliferation of a wide variety of tumor cells; downregulate transcription factors NF-κB, AP-1, and Egr-1; downregulate the expression of COX-2, LOX, iNOS, MMP-9, uPA, TNF, chemokines, cell surface adhesion molecules, and cyclin D1; downregulate growth factor receptors (such as EGFR and HER2); and inhibit the activity of c-Jun N-terminal kinase, protein tyrosine kinases, and protein serine/threonine kinases. In several systems, curcumin is a potent antioxidant and anti-inflammatory agent.
Further evidence suggests that curcumin can suppress tumor initiation, promotion, and metastasis. Human clinical trials have indicated no dose-limiting toxicity when curcumin is administered at doses up to 10 grams per day. All these studies suggest that curcumin has enormous potential in the prevention and treatment of cancer.
3. Resveratrol (Vitis vinifera)
The history of resveratrol can be traced back thousands of years. Perhaps the first known use of grape extracts for human health occurred > 2000 years ago in ‘darakchasava,’ a well-known Indian herbal preparation whose main ingredient is Vitis vinifera L. This ‘Ayurvedic’ medicine is prescribed as a cardiotonic and also is given for other disorders [61]. The use of dried grapes (also called manakka) as a cardiotonic is well documented. High-performance liquid chromatography analysis of darakchasava revealed the presence of polyphenols, such as resveratrol and pterostilbene. Interest in this age-old formulation grew in light of this new knowledge of resveratrol. Resveratrol, trans-3,5,4′-trihydroxy stilbene, was first isolated in 1940 as a constituent of the roots of white hellebore (Veratrum grandiflorum O. Loes) but has since been found in various plants, including grapes, berries, and peanuts [62-65]. Besides cardioprotective effects, resveratrol exhibits anticancer properties, as suggested by its ability to suppress proliferation of a wide variety of tumor cells, including lymphoid and myeloid cancers, multiple myeloma, cancers of the breast, prostate, stomach, colon, pancreas, and thyroid, melanoma, head, and neck squamous cell carcinomas, ovarian carcinoma and cervical carcinoma. The growth-inhibitory effects of resveratrol are mediated through cell-cycle arrest, upregulation of p21Cip1/WAF1, p53 and Bax, downregulation of survivin, cyclin D1, cyclin E, bcl-2, bcl-XL, and cIAPs, and activation of caspases. Resveratrol has been shown to suppress the activation of several transcription factors, including NF-κB, AP-1, and Egr-1; inhibit protein kinases, including IκBα kinase, JNK, MAPK, Akt, PKC, PKD, and casein kinase II; and downregulate products of genes such as COX-2, 5-lipoxygenase (5-LOX), VEGF, IL-1, IL-6, IL-8,
androgen receptor, and, prostate-specific antigen. These activities account for this stilbene’s suppression of angiogenesis. Resveratrol also has been shown to potentiate the apoptotic effects of cytokines (such as TRAIL, chemotherapeutic agents and γ-radiation. Pharmacokinetic studies have revealed that resveratrol’s target organs are liver and kidney, where it is concentrated after absorption and is mainly converted to a sulfated form and a glucuronide conjugate. In vivo, resveratrol blocks the multistep process of carcinogenesis at various stages: It blocks carcinogen activation by inhibiting aryl hydrocarbon-induced CYP1A1 expression and activity and suppresses tumor initiation, promotion, and progression. Besides chemopreventive effects, resveratrol appears to exhibit therapeutic effects against cancer. Limited data in humans have revealed that resveratrol is pharmacologically quite safe. Currently, structural analogs of resveratrol with improved bioavailability are being pursued as potential therapeutic agents for cancer.
4.Flavopiridol (Dysoxylum binectariferum)
4.Flavopiridol (Dysoxylum binectariferum)
4.Flavopiridol (Dysoxylum binectariferum)
Flavopiridol is a semisynthetic flavonoid closely related to a compound originally isolated from the stem bark of Dysoxylum binectariferum (also called rohitukine from Amoora rohituka), a plant indigenous to India and described in Ayurveda. The parent compound is identical to flavopiridol except that a methyl group replaces the chlorophenyl moiety at position 2. Flavopiridol is a potent inhibitor of cyclin-dependent kinase (CDK) 1, CDK 2, CDK 4 and CDK 7 [66]. It inhibits CDKs by competing with adenosine triphosphate at the nucleotide-binding site on CDKs, as indicated by kinetics studies [67] and X-ray crystallography of the CDK 2–flavopiridol complex [68]. The tyrosine phosphorylation of CDK 2 is also inhibited by this flavone[69]. Through inhibition of CDKs, flavopiridol induces arrest of cell growth at the G1 and G2 phases of the cell cycle [66,70]. Because of its ability to suppress the growth of breast carcinoma [66], lung carcinoma [71], chronic B cell leukemia and lymphoma [72-74], multiple myeloma [75] and head and neck squamous cell carcinoma [76], flavopiridol is currently in clinical trials for the treatment of several cancers [77-79]. Flavopiridol also has been shown to enhance the activity of other growth-suppressing agents, such as TNF, doxorubicin, and etoposide [80-84]. Flavopiridol also inhibits CDKs, induces apoptosis, suppresses inflammation, and modulates the immune response. Flavopiridol suppressed TNF activation of NF-κB in a dose- and time-dependent manner in several cell types, with optimal inhibition occurring when cells were treated with 100 nM of flavopiridol for 6 h [85].
5. Zerumbone (Zingiber zerumbet Smith)
4.Flavopiridol (Dysoxylum binectariferum)
4.Flavopiridol (Dysoxylum binectariferum)
Zerumbone (2,6,9,9-tetramethyl-[2E,6E,10E]-cycloundeca- 2,6,10-tried-1-one) was first isolated in 1956 from the essential oil of the rhizomes of wild ginger, Zingiber zerumbet Smith, which is widespread in Southeast Asia [86,87]. Over the years, a wide variety of activities have been ascribed to this compound [88-94]. For instance, zerumbone has been found to suppress the proliferation of colon cancer [93,94] and breast cancer [93], with minimal effects on normal cells [94]. Zerumbone also has been shown to suppress inflammation [92], suppress the initiation and promotion of skin tumors in mice [91], and prevent azoxymethane-induced aberrant crypt foci formation in rats [90]. Besides, this terpenoid has been shown to suppress dextran sodium sulfate-induced colitis in mice [95] and to inhibit the activation of the phorbol ester-induced Epstein-Barr virus [88]. Zerumbone also has been found to suppress superoxide and nitric oxide generation [89] and downregulate COX-2 [96], IL-1β [95] and TNF [94,95]. A potential explanation for several of these activities is that zerumbone may downregulate NF-κB activation [97]. The authors’ laboratory has shown that zerumbone suppressed NF-κB activation induced by a variety of agents. Interestingly, α-humulene, a structural analog of zerumbone lacking the carbonyl group, was completely inactive. Besides being inducible, constitutively active NF-κB also was inhibited. This downregulation potentiated apoptosis induced by cytokines and chemotherapeutic agents. Zerumbone’s inhibition of the expression of these NF-κB-regulated genes also correlated with the suppression of TNF-induced invasion activity. Overall, this inhibition may provide a molecular basis for exploring zerumbone’s potential in the prevention and treatment of cancer.
6. Withanolide (Withania sominifera)
7. Boswellic acid (Boswellia serrata)
7. Boswellic acid (Boswellia serrata)
The medicinal plant Withania somnifera is widely known for its anti-inflammatory, cardioactive and CNS effects. In Ayurveda, withanolide, which is extracted from W. somnifera, is employed in the treatment of arthritis and menstrual disorders and is known to be potent inhibitors of angiogenesis, inflammation, tumor development, metastasis and oxidative stress, and a promoter of cardioprotection. Many pharmacological studies have investigated the properties of W. foraminifera in an attempt to authenticate its use as a multipurpose medical agent. Experimental studies have shown that W. foraminifera possesses anti-inflammatory, antitumor, cardioprotective, and antioxidant properties. Withaferin A, one of the compounds in the withanolide family, is a potent inhibitor of angiogenesis. It also appears to exert a positive influence on the endocrine, urogenital, and central nervous systems. In recent years, herbal formulations containing substantial amounts of W. foraminifera root extract have been evaluated in small clinical trials and shown to have efficacy in the treatment of osteoarthritis. Excepts are also known to inhibit tumor growth in vivo significantly. However, the mechanisms responsible for the antitumor effects of withanolide are still unknown. The authors found that withanolide suppressed NF-κB activation induced by a wide variety of inflammatory and carcinogenic agents, including TNF, IL-1β, doxorubicin, and cigarette smoke condensate. Withanolide also enhanced the apoptosis induced by TNF and chemotherapeutic agents and suppressed invasion. These results indicate that withanolide inhibits activation of NF-κB and NF-κB-regulated gene expression. This may explain their ability to enhance apoptosis and inhibit invasion.
7. Boswellic acid (Boswellia serrata)
7. Boswellic acid (Boswellia serrata)
7. Boswellic acid (Boswellia serrata)
Boswellic acid (BA) is an active component of Boswellia serrata (also known as Salai guggul). The gum-resin of this plant is used in Ayurvedic medicine to treat rheumatic diseases, respiratory diseases, and liver disorders [98-100]. Extensive research within the last 30 years has identified the active component of this resin as BA (a pentacyclic triterpenic acid) and its derivatives (acetyl-β-boswellic acid, 11-keto-β-boswellic acid and acetyl-11-keto-β-boswellic acid [AKBA]) [101,102]. The traditional therapeutic usefulness of BA is a result of its anti-inflammatory activity, possibly mediated through the inhibition of 5-LOX [102-104] and leukocyte elastase [105,106]. In animal models of inflammation, BA is effective against Crohn’s disease, ulcerative colitis, and ileitis [107-109]; adjuvant or BSA-induced arthritis [110,111]; galactosamine/endotoxin-induced hepatitis in mice [112]; and osteoarthritis [113]. BA has antitumor effects in addition to its anti-inflammatory effects. It has been found to have activity against brain tumors [114,115], leukemic cells [116,117], colon cancer cells [118], metastatic melanoma and fibrosarcoma cells [119], and hepatoma [118]. BA also has been shown to inhibit the azoxymethane-induced formation of aberrant crypt foci in the colon of mice [120]. AKBA, a component of Boswellia serrata, is a pentacyclic terpenoid that is active against numerous inflammatory diseases, including cancer, arthritis, chronic colitis, ulcerative colitis, Crohn’s disease, and bronchial asthma. The authors found that AKBA potentiates the apoptosis induced by TNF and chemotherapeutic agents suppress TNF-induced invasion and inhibits the receptor activator of NF-κB ligand-induced osteoclastogenesis, all of which are known to require NF-κB activation (Takada et al., unpublished observations, 2005).
8. Fruits and vegetables

Fruits and vegetables are an integral part of Ayurvedic medicine. Steinmetz and Potter reviewed the scientific literature on the relationship between vegetable and fruit consumption and the risk of cancer [121]. After reviewing results from 206 human epidemiological studies and 22 animal studies, they found clear evidence that a higher intake of vegetables and fruits protect against cancers of the stomach, esophagus, lung, oral cavity and pharynx, endometrium, pancreas and colon. The types of vegetables and fruits that most often appear to be protective against cancer are raw vegetables, followed by cooked allium vegetables, carrots, green vegetables, cruciferous vegetables and tomatoes. The substances in vegetables and fruits that may help protect against cancer include dithiolthiones, isothiocyanates, indole-3-carbinol (I3C), allium compounds, isoflavones, protease inhibitors, saponins, phytosterols, inositol hexaphosphate, vitamin C, d-limonene, lutein, folic acid, β-carotene, lycopene, selenium, vitamin E and dietary fiber. How fruits and vegetables mediate their effects is beginning to be revealed [122,123]. For instance, I3C is produced by members of the family Cruciferae, and particularly members of the genus Brassica (for example, cabbage, radishes, cauliflower, broccoli, Brussels sprouts, and daikon). Under acidic conditions, I3C is converted to a series of oligomeric products (among which 3,3’-diindolylmethane is a significant component) believed to be responsible for its biological effects in vivo. In vitro, I3C has been shown to suppress the proliferation of various tumor cells, including those from breast, prostate, endometrial, and colon cancers and leukemia; induce G1/S cell-cycle arrest, and induce apoptosis. The cell-cycle arrest involves downregulation of cyclin D1, cyclin E, CDK2, CDK4 and CDK6 and upregulation of p15, p21, and p27. Apoptosis by I3C involves downregulation of antiapoptotic gene products, including bcl-2, bcl-XL, survivin, IAP, X-linked inhibitor of apoptosis, and Fas-associated death domain protein-like IL-1-β-converting enzyme inhibitory protein (FLIP), upregulation of pro-apoptotic protein bax, the release of mitochondrial cytochrome c, and activation of caspase-9 and caspase-3. This agent inhibits the activation of various transcription factors, including NF-κB, SP1, estrogen receptor, androgen receptor, and nuclear factor-E2-related factor 2. This indole potentiates the effects of TRAIL through induction of death receptors and synergizes with chemotherapeutic agents through the downregulation of P-gp. In vivo, I3C was found to be a potent chemopreventive agent for hormone-dependent cancers, such as breast and cervical cancers. These effects are mediated through its ability to induce apoptosis, inhibit DNA-carcinogen adduct formation, suppress free radical production, stimulate 2-hydroxylation of estradiol, and inhibit invasion and angiogenesis. Numerous studies have indicated that I3C also has intense hepatoprotective activity against various carcinogens. Initial clinical trials in women have shown that I3C is a promising agent against breast and cervical cancers.
Expert opinion
The biology of cancer is much better understood today than it was a few decades ago. Despite this increasing knowledge, the incidence of cancer is higher today than it was 30 years ago. Epidemiology has revealed that certain diseases are more common among people of some cultures than others. Cancers of the lung, colon, prostate, and breast are prevalent in Western countries; they are not as commonplace in Eastern countries. Similarly, diseases of the head and neck and the cervix are most common in India, whereas stomach cancer is most prevalent in Japan. Migration from native to the adopted country, however, exposes an individual to the same cancer risk and incidence as that of others living in the chosen country. This phenomenon suggests a minimal role for genotype and a more significant part of a lifestyle. These findings have prompted the US National Cancer Institute to examine the traditional concepts of lifestyle that play a role in cancer prevention. Ayurveda is an intricate system of healing that originated in India thousands of years ago. Historical evidence of Ayurveda can be found in the ancient books of wisdom known as the Vedas that were written over 6000 years ago. Ayurveda provides novel approaches to cancer prevention that are considered safe. Ayurvedic treatment of cancer involves prevention, surgical removal of tumors, herbal remedies, dietary modifications, and spiritual therapies (e.g., detoxification, rejuvenation, prayer, music therapy, aromatherapy, gem therapy, sound therapy, stress relief, meditation, yoga, and astrology). The current emphasis should be laid on the identification of the mechanism of action of ayurvedic drugs and the prevention and treatment of cancer by combining these treatments with modern developments in medicine.